2022
Kang J, Nathan A, Weinand K, Zhang F, Millard N, Rumker L, Moody DB, Korsunsky I, Raychaudhuri S.
Efficient and precise single-cell reference atlas mapping with Symphony [Internet]. Nature Communications 2022;12(5890)
Publisher's VersionAbstractRecent advances in single-cell technologies and integration algorithms make it possible to construct comprehensive reference atlases encompassing many donors, studies, disease states, and sequencing platforms. Much like mapping sequencing reads to a reference genome, it is essential to be able to map query cells onto complex, multimillion-cell reference atlases to rapidly identify relevant cell states and phenotypes. We present Symphony (
https://github.com/immunogenomics/symphony), an algorithm for building large-scale, integrated reference atlases in a convenient, portable format that enables efficient query mapping within seconds. Symphony localizes query cells within a stable low-dimensional reference embedding, facilitating reproducible downstream transfer of reference-defined annotations to the query. We demonstrate the power of Symphony in multiple real-world datasets, including (1) mapping a multi-donor, multi-species query to predict pancreatic cell types, (2) localizing query cells along a developmental trajectory of fetal liver hematopoiesis, and (3) inferring surface protein expression with a multimodal CITE-seq atlas of memory T cells.
Lopez BGC, Kohale IN, Du Z, Korsunsky I, Abdelmoula WM, Dai Y, Stopka SA, Gaglia G, Randall EC, Regan MS, Basu SS, Clark AR, Marin B-M, Mladek AC, Burgenske DM, Agar JN, Supko JG, Grossman SA, Nabors LB, Raychaudhuri S, Ligon KL, Wen PY, Alexander B, Lee EQ, Santagata S, Sarkaria J, White FM, Agar NYR.
Multimodal platform for assessing drug distribution and response in clinical trials. Neuro Oncol 2022;24(1):64-77.
Abstract
BACKGROUND: Response to targeted therapy varies between patients for largely unknown reasons. Here, we developed and applied an integrative platform using mass spectrometry imaging (MSI), phosphoproteomics, and multiplexed tissue imaging for mapping drug distribution, target engagement, and adaptive response to gain insights into heterogeneous response to therapy.
METHODS: Patient-derived xenograft (PDX) lines of glioblastoma were treated with adavosertib, a Wee1 inhibitor, and tissue drug distribution was measured with MALDI-MSI. Phosphoproteomics was measured in the same tumors to identify biomarkers of drug target engagement and cellular adaptive response. Multiplexed tissue imaging was performed on sister sections to evaluate spatial co-localization of drug and cellular response. The integrated platform was then applied on clinical specimens from glioblastoma patients enrolled in the phase 1 clinical trial.
RESULTS: PDX tumors exposed to different doses of adavosertib revealed intra- and inter-tumoral heterogeneity of drug distribution and integration of the heterogeneous drug distribution with phosphoproteomics and multiplexed tissue imaging revealed new markers of molecular response to adavosertib. Analysis of paired clinical specimens from patients enrolled in the phase 1 clinical trial informed the translational potential of the identified biomarkers in studying patient's response to adavosertib.
CONCLUSIONS: The multimodal platform identified a signature of drug efficacy and patient-specific adaptive responses applicable to preclinical and clinical drug development. The information generated by the approach may inform mechanisms of success and failure in future early phase clinical trials, providing information for optimizing clinical trial design and guiding future application into clinical practice.
Lagattuta KA, Kang JB, Nathan A, Pauken KE, Jonsson AH, Rao DA, Sharpe AH, Ishigaki K, Raychaudhuri S.
Repertoire analyses reveal T cell antigen receptor sequence features that influence T cell fate. Nat Immunol 2022;23(3):446-457.
AbstractT cells acquire a regulatory phenotype when their T cell antigen receptors (TCRs) experience an intermediate- to high-affinity interaction with a self-peptide presented via the major histocompatibility complex (MHC). Using TCRβ sequences from flow-sorted human cells, we identified TCR features that promote regulatory T cell (Treg) fate. From these results, we developed a scoring system to quantify TCR-intrinsic regulatory potential (TiRP). When applied to the tumor microenvironment, TiRP scoring helped to explain why only some T cell clones maintained the conventional T cell (Tconv) phenotype through expansion. To elucidate drivers of these predictive TCR features, we then examined the two elements of the Treg TCR ligand separately: the self-peptide and the human MHC class II molecule. These analyses revealed that hydrophobicity in the third complementarity-determining region (CDR3β) of the TCR promotes reactivity to self-peptides, while TCR variable gene (TRBV gene) usage shapes the TCR's general propensity for human MHC class II-restricted activation.
Pauken KE, Lagattuta KA, Lu BY, Lucca LE, Daud AI, Hafler DA, Kluger HM, Raychaudhuri S, Sharpe AH.
TCR-sequencing in cancer and autoimmunity: barcodes and beyond. Trends Immunol 2022;43(3):180-194.
AbstractThe T cell receptor (TCR) endows T cells with antigen specificity and is central to nearly all aspects of T cell function. Each naïve T cell has a unique TCR sequence that is stably maintained during cell division. In this way, the TCR serves as a molecular barcode that tracks processes such as migration, differentiation, and proliferation of T cells. Recent technological advances have enabled sequencing of the TCR from single cells alongside deep molecular phenotypes on an unprecedented scale. In this review, we discuss strengths and limitations of TCR sequences as molecular barcodes and their application to study immune responses following Programmed Death-1 (PD-1) blockade in cancer. Additionally, we consider applications of TCR data beyond use as a barcode.
Reshef Y, Rumker L, Kang JB, Nathan A, Korsunsky I, Asgari S, Murray MB, Moody BD, Raychaudhuri S.
Co-varying neighborhood analysis identifies cell populations associated with phenotypes of interest from single-cell transcriptomics. Nat Biotechnol 2022;40(3):355-363.
AbstractAs single-cell datasets grow in sample size, there is a critical need to characterize cell states that vary across samples and associate with sample attributes, such as clinical phenotypes. Current statistical approaches typically map cells to clusters and then assess differences in cluster abundance. Here we present co-varying neighborhood analysis (CNA), an unbiased method to identify associated cell populations with greater flexibility than cluster-based approaches. CNA characterizes dominant axes of variation across samples by identifying groups of small regions in transcriptional space-termed neighborhoods-that co-vary in abundance across samples, suggesting shared function or regulation. CNA performs statistical testing for associations between any sample-level attribute and the abundances of these co-varying neighborhood groups. Simulations show that CNA enables more sensitive and accurate identification of disease-associated cell states than a cluster-based approach. When applied to published datasets, CNA captures a Notch activation signature in rheumatoid arthritis, identifies monocyte populations expanded in sepsis and identifies a novel T cell population associated with progression to active tuberculosis.
Ishigaki K, Lagattuta KA, Luo Y, James EA, Buckner JH, Raychaudhuri S.
HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors. Nat Genet 2022;54(4):393-402.
AbstractPolymorphisms in the human leukocyte antigen (HLA) genes strongly influence autoimmune disease risk. HLA risk alleles may influence thymic selection to increase the frequency of T cell receptors (TCRs) reactive to autoantigens (central hypothesis). However, research in human autoimmunity has provided little evidence supporting the central hypothesis. Here we investigated the influence of HLA alleles on TCR composition at the highly diverse complementarity determining region 3 (CDR3), which confers antigen recognition. We observed unexpectedly strong HLA-CDR3 associations. The strongest association was found at HLA-DRB1 amino acid position 13, the position that mediates genetic risk for multiple autoimmune diseases. We identified multiple CDR3 amino acid features enriched by HLA risk alleles. Moreover, the CDR3 features promoted by the HLA risk alleles are more enriched in candidate pathogenic TCRs than control TCRs (for example, citrullinated epitope-specific TCRs in patients with rheumatoid arthritis). Together, these results provide genetic evidence supporting the central hypothesis.
Guan S, Mehta B, Slater D, Thompson JR, DiCarlo E, Pannellini T, Pearce-Fisher D, Zhang F, Raychaudhuri S, Hale C, Jiang CS, Goodman S, Orange DE.
Rheumatoid Arthritis Synovial Inflammation Quantification Using Computer Vision. ACR Open Rheumatol 2022;4(4):322-331.
AbstractOBJECTIVE: We quantified inflammatory burden in rheumatoid arthritis (RA) synovial tissue by using computer vision to automate the process of counting individual nuclei in hematoxylin and eosin images.
METHODS: We adapted and applied computer vision algorithms to quantify nuclei density (count of nuclei per unit area of tissue) on synovial tissue from arthroplasty samples. A pathologist validated algorithm results by labeling nuclei in synovial images that were mislabeled or missed by the algorithm. Nuclei density was compared with other measures of RA inflammation such as semiquantitative histology scores, gene-expression data, and clinical measures of disease activity.
RESULTS: The algorithm detected a median of 112,657 (range 8,160-821,717) nuclei per synovial sample. Based on pathologist-validated results, the sensitivity and specificity of the algorithm was 97% and 100%, respectively. The mean nuclei density calculated by the algorithm was significantly higher (P < 0.05) in synovium with increased histology scores for lymphocytic inflammation, plasma cells, and lining hyperplasia. Analysis of RNA sequencing identified 915 significantly differentially expressed genes in correlation with nuclei density (false discovery rate is less than 0.05). Mean nuclei density was significantly higher (P < 0.05) in patients with elevated levels of C-reactive protein, erythrocyte sedimentation rate, rheumatoid factor, and cyclized citrullinated protein antibody.
CONCLUSION: Nuclei density is a robust measurement of inflammatory burden in RA and correlates with multiple orthogonal measurements of inflammation.
Maurits MP, Korsunsky I, Raychaudhuri S, Murphy SN, Smoller JW, Weiss ST, Huizinga TWJ, Reinders MJT, Karlson EW, van den Akker EB, Knevel R.
A framework for employing longitudinally collected multicenter electronic health records to stratify heterogeneous patient populations on disease history. J Am Med Inform Assoc 2022;29(5):761-769.
AbstractOBJECTIVE: To facilitate patient disease subset and risk factor identification by constructing a pipeline which is generalizable, provides easily interpretable results, and allows replication by overcoming electronic health records (EHRs) batch effects.
MATERIAL AND METHODS: We used 1872 billing codes in EHRs of 102 880 patients from 12 healthcare systems. Using tools borrowed from single-cell omics, we mitigated center-specific batch effects and performed clustering to identify patients with highly similar medical history patterns across the various centers. Our visualization method (PheSpec) depicts the phenotypic profile of clusters, applies a novel filtering of noninformative codes (Ranked Scope Pervasion), and indicates the most distinguishing features.
RESULTS: We observed 114 clinically meaningful profiles, for example, linking prostate hyperplasia with cancer and diabetes with cardiovascular problems and grouping pediatric developmental disorders. Our framework identified disease subsets, exemplified by 6 "other headache" clusters, where phenotypic profiles suggested different underlying mechanisms: migraine, convulsion, injury, eye problems, joint pain, and pituitary gland disorders. Phenotypic patterns replicated well, with high correlations of ≥0.75 to an average of 6 (2-8) of the 12 different cohorts, demonstrating the consistency with which our method discovers disease history profiles.
DISCUSSION: Costly clinical research ventures should be based on solid hypotheses. We repurpose methods from single-cell omics to build these hypotheses from observational EHR data, distilling useful information from complex data.
CONCLUSION: We establish a generalizable pipeline for the identification and replication of clinically meaningful (sub)phenotypes from widely available high-dimensional billing codes. This approach overcomes datatype problems and produces comprehensive visualizations of validation-ready phenotypes.
Fava A, Rao DA, Mohan C, Zhang T, Rosenberg A, Fenaroli P, Belmont MH, Izmirly P, Clancy R, Trujillo JM, Fine D, Arazi A, Berthier CC, Davidson A, James JA, Diamond B, Hacohen N, Wofsy D, Raychaudhuri S, Apruzzese W, Buyon J, Petri M.
Urine Proteomics and Renal Single-Cell Transcriptomics Implicate Interleukin-16 in Lupus Nephritis. Arthritis Rheumatol 2022;74(5):829-839.
AbstractOBJECTIVE: Current lupus nephritis (LN) treatments are effective in only 30% of patients, emphasizing the need for novel therapeutic strategies. We undertook this study to develop mechanistic hypotheses and explore novel biomarkers by analyzing the longitudinal urinary proteomic profiles in LN patients undergoing treatment.
METHODS: We quantified 1,000 urinary proteins in 30 patients with LN at the time of the diagnostic renal biopsy and after 3, 6, and 12 months. The proteins and molecular pathways detected in the urine proteome were then analyzed with respect to baseline clinical features and longitudinal trajectories. The intrarenal expression of candidate biomarkers was evaluated using single-cell transcriptomics of renal biopsy sections from LN patients.
RESULTS: Our analysis revealed multiple biologic pathways, including chemotaxis, neutrophil activation, platelet degranulation, and extracellular matrix organization, which could be noninvasively quantified and monitored in the urine. We identified 237 urinary biomarkers associated with LN, as compared to controls without systemic lupus erythematosus. Interleukin-16 (IL-16), CD163, and transforming growth factor β mirrored intrarenal nephritis activity. Response to treatment was paralleled by a reduction in urinary IL-16, a CD4 ligand with proinflammatory and chemotactic properties. Single-cell RNA sequencing independently demonstrated that IL16 is the second most expressed cytokine by most infiltrating immune cells in LN kidneys. IL-16-producing cells were found at key sites of kidney injury.
CONCLUSION: Urine proteomics may profoundly change the diagnosis and management of LN by noninvasively monitoring active intrarenal biologic pathways. These findings implicate IL-16 in LN pathogenesis, designating it as a potentially treatable target and biomarker.
Mysore V, Tahir S, Furuhashi K, Arora J, Rosetti F, Cullere X, Yazbeck P, Sekulic M, Lemieux ME, Raychaudhuri S, Horwitz BH, Mayadas TN.
Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J Exp Med 2022;219(5)
AbstractMonocytes undergo phenotypic and functional changes in response to inflammatory cues, but the molecular signals that drive different monocyte states remain largely undefined. We show that monocytes acquire macrophage markers upon glomerulonephritis and may be derived from CCR2+CX3CR1+ double-positive monocytes, which are preferentially recruited, dwell within glomerular capillaries, and acquire proinflammatory characteristics in the nephritic kidney. Mechanistically, the transition to immature macrophages begins within the vasculature and relies on CCR2 in circulating cells and TNFR2 in parenchymal cells, findings that are recapitulated in vitro with monocytes cocultured with TNF-TNFR2-activated endothelial cells generating CCR2 ligands. Single-cell RNA sequencing of cocultures defines a CCR2-dependent monocyte differentiation path associated with the acquisition of immune effector functions and generation of CCR2 ligands. Immature macrophages are detected in the urine of lupus nephritis patients, and their frequency correlates with clinical disease. In conclusion, CCR2-dependent functional specialization of monocytes into macrophages begins within the TNF-TNFR2-activated vasculature and may establish a CCR2-based autocrine, feed-forward loop that amplifies renal inflammation.
Nathan A, Asgari S, Ishigaki K, Valencia C, Amariuta T, Luo Y, Beynor JI, Baglaenko Y, Suliman S, Price AL, Lecca L, Murray MB, Moody BD, Raychaudhuri S.
Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 2022;606(7912):120-128.
AbstractNon-coding genetic variants may cause disease by modulating gene expression. However, identifying these expression quantitative trait loci (eQTLs) is complicated by differences in gene regulation across fluid functional cell states within cell types. These states-for example, neurotransmitter-driven programs in astrocytes or perivascular fibroblast differentiation-are obscured in eQTL studies that aggregate cells1,2. Here we modelled eQTLs at single-cell resolution in one complex cell type: memory T cells. Using more than 500,000 unstimulated memory T cells from 259 Peruvian individuals, we show that around one-third of 6,511 cis-eQTLs had effects that were mediated by continuous multimodally defined cell states, such as cytotoxicity and regulatory capacity. In some loci, independent eQTL variants had opposing cell-state relationships. Autoimmune variants were enriched in cell-state-dependent eQTLs, including risk variants for rheumatoid arthritis near ORMDL3 and CTLA4; this indicates that cell-state context is crucial to understanding potential eQTL pathogenicity. Moreover, continuous cell states explained more variation in eQTLs than did conventional discrete categories, such as CD4+ versus CD8+, suggesting that modelling eQTLs and cell states at single-cell resolution can expand insight into gene regulation in functionally heterogeneous cell types.
Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, Taylor E, Ravindran R, Kemble S, Watts GFM, Jonsson HA, Jeong Y, Athar H, Windell D, Kang JB, Friedrich M, Turner J, Nayar S, Fisher BA, Raza K, Marshall JL, Croft AP, Tamura T, Sholl LM, Vivero M, Rosas IO, Bowman SJ, Coles M, Frei AP, Lassen K, Filer A, Powrie F, Buckley CD, Brenner MB, Raychaudhuri S.
Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med (N Y) 2022;3(7):481-518.e14.
AbstractBACKGROUND: Pro-inflammatory fibroblasts are critical for pathogenesis in rheumatoid arthritis, inflammatory bowel disease, interstitial lung disease, and Sjögren's syndrome and represent a novel therapeutic target for chronic inflammatory disease. However, the heterogeneity of fibroblast phenotypes, exacerbated by the lack of a common cross-tissue taxonomy, has limited our understanding of which pathways are shared by multiple diseases.
METHODS: We profiled fibroblasts derived from inflamed and non-inflamed synovium, intestine, lungs, and salivary glands from affected individuals with single-cell RNA sequencing. We integrated all fibroblasts into a multi-tissue atlas to characterize shared and tissue-specific phenotypes.
FINDINGS: Two shared clusters, CXCL10+CCL19+ immune-interacting and SPARC+COL3A1+ vascular-interacting fibroblasts, were expanded in all inflamed tissues and mapped to dermal analogs in a public atopic dermatitis atlas. We confirmed these human pro-inflammatory fibroblasts in animal models of lung, joint, and intestinal inflammation.
CONCLUSIONS: This work represents a thorough investigation into fibroblasts across organ systems, individual donors, and disease states that reveals shared pathogenic activation states across four chronic inflammatory diseases.
FUNDING: Grant from F. Hoffmann-La Roche (Roche) AG.
Asgari S, Luo Y, Huang C-C, Zhang Z, Calderon R, Jimenez J, Yataco R, Contreras C, Galea JT, Lecca L, Jones D, Moody BD, Murray MB, Raychaudhuri S.
Higher native Peruvian genetic ancestry proportion is associated with tuberculosis progression risk. Cell Genom 2022;2(7)
AbstractWe investigated whether ancestry-specific genetic factors affect tuberculosis (TB) progression risk in a cohort of admixed Peruvians. We genotyped 2,105 patients with TB and 1,320 household contacts (HHCs) who were infected with Mycobacterium tuberculosis (M. tb) but did not develop TB and inferred each individual's proportion of native Peruvian genetic ancestry. Our HHC study design and our data on potential confounders allowed us to demonstrate increased risk independent of socioeconomic factors. A 10% increase in individual-level native Peruvian genetic ancestry proportion corresponded to a 25% increased TB progression risk. This corresponds to a 3-fold increased risk for individuals in the highest decile of native Peruvian genetic ancestry versus the lowest decile, making native Peruvian genetic ancestry comparable in effect to clinical factors such as diabetes. Our results suggest that genetic ancestry is a major contributor to TB progression risk and highlight the value of including diverse populations in host genetic studies.
Deakin CT, Bowes J, Rider LG, Miller FW, Pachman LM, Sanner H, Rouster-Stevens K, Mamyrova G, Curiel R, Feldman BM, Huber AM, Reed AM, Schmeling H, Cook CG, Marshall LR, Wilkinson MLIG, Eyre S, Raychaudhuri S, Wedderburn LR, and the Juvenile Dermatomyositis Cohort and Biomarker Study, the Childhood Myositis Heterogeneity Study Group MG.
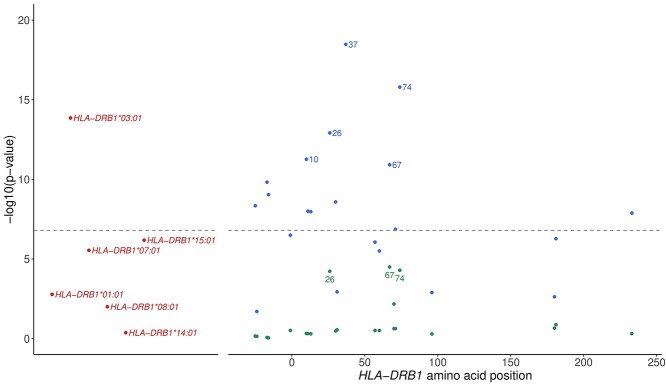
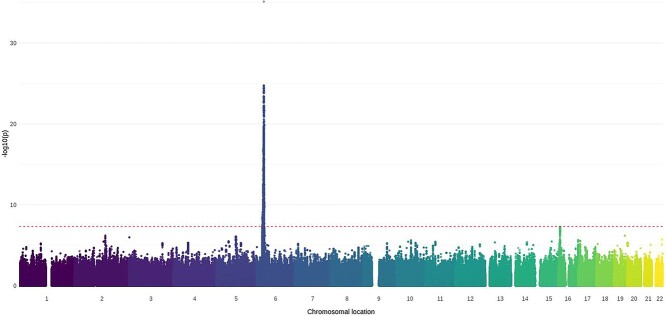
Association with HLA-DRβ1 position 37 distinguishes juvenile dermatomyositis from adult-onset myositis. Human Molecular Genetics 2022;31(14):2471–2481.
Abstract
Juvenile dermatomyositis (JDM) is a rare, severe autoimmune disease and the most common idiopathic inflammatory myopathy of children. JDM and adult-onset dermatomyositis (DM) have similar clinical, biological and serological features, although these features differ in prevalence between childhood-onset and adult-onset disease, suggesting that age of disease onset may influence pathogenesis. Therefore, a JDM-focused genetic analysis was performed using the largest collection of JDM samples to date. Caucasian JDM samples (n = 952) obtained via international collaboration were genotyped using the Illumina HumanCoreExome chip. Additional non-assayed human leukocyte antigen (HLA) loci and genome-wide single-nucleotide polymorphisms (SNPs) were imputed. HLA-DRB1*03:01 was confirmed as the classical HLA allele most strongly associated with JDM [odds ratio (OR) 1.66; 95% confidence interval (CI) 1.46, 1.89; P = 1.4 × 10-14], with an independent association at HLA-C*02:02 (OR = 1.74; 95% CI 1.42, 2.13, P = 7.13 × 10-8). Analyses of amino acid positions within HLA-DRB1 indicated that the strongest association was at position 37 (omnibus P = 3.3 × 10-19), with suggestive evidence this association was independent of position 74 (omnibus P = 5.1 × 10-5), the position most strongly associated with adult-onset DM. Conditional analyses also suggested that the association at position 37 of HLA-DRB1 was independent of some alleles of the Caucasian HLA 8.1 ancestral haplotype (AH8.1) such as HLA-DQB1*02:01 (OR = 1.62; 95% CI 1.36, 1.93; P = 8.70 × 10-8), but not HLA-DRB1*03:01 (OR = 1.49; 95% CR 1.24, 1.80; P = 2.24 × 10-5). No associations outside the HLA region were identified. Our findings confirm previous associations with AH8.1 and HLA-DRB1*03:01, HLA-C*02:02 and identify a novel association with amino acid position 37 within HLA-DRB1, which may distinguish JDM from adult DM.
 Figure 1 Figure 2 Figure 3
Figure 1 Figure 2 Figure 3 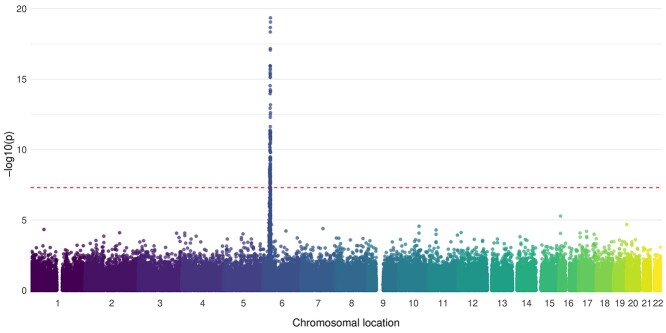{kind=link}
{kind=link}
{kind=link}